CITE-seq - normalization with CLR
Why CLR?
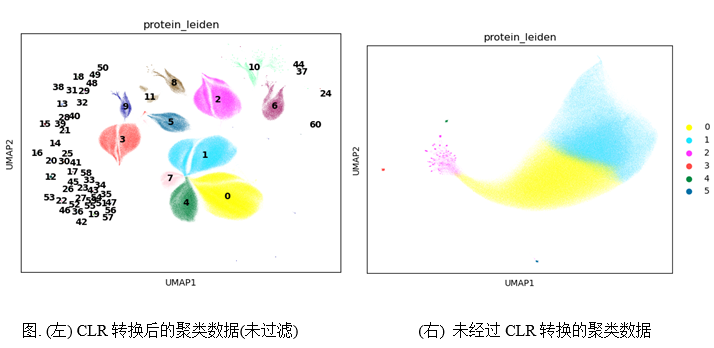
问题表述: 尽管不同的蛋白表达量之间具有差异性,但clusters之间无法较好分离
主要原因: 在对转录组+蛋白组的原始数据预处理(pre-processing)部分,RNA filtering流程无较大差异,但基本上所有CITE-seq文章中对于蛋白组数据在聚类前都使用了CLR normalization方法。
由于对于给定单一细胞,每个ADT count都可以被认为是一个整体(all ADT counts)的一个部分,且存在非特异性结合(这也是蛋白数据存在大量噪声的原因之一),更倾向于将ADT-tag counts数据认为是组成性数据(compositional data)。而传统统计学分析并不适用于这类数据(原因在组成分析的创始论文有具体阐述Aitchison 1989),可简单理解为由于组成性数据更关注相对量而不是绝对值的大小,且总量受限制(在CITE-Seq语境下,即考虑到目前可选择的蛋白marker种类限制,应更关注不同蛋白间的表达量差异而非具体counts的多少),不同ADT-tags counts相当于限制性变量,不同于常用的非限制性变量(unconstrained random variables)。因此使用CLR对数据进行转换应当是必要的。
- CLR-transformed ADT vector y for each cell where
and x is the vector of ADT counts including one pesudocount for each component, and g(x) if the geometric mean of x.
对于CLR修正前后的数据进行了聚类比较,使用了相同的参数resolution=0.1。