MERFISH - overview
背景
1998年出现的单分子荧光原位杂交(smFISH)技术可量化mRNA分子拷贝数和空间位置。smFISH技术利用短寡核苷酸探针靶向检测转录本,当多条寡核苷酸与同一转录本结合,产生足够强度的荧光信号后进行定量。但通过smFISH同时测量的RNA种类数量有限,为解决通量和多轮成像准确性问题,Prof. Xiaowei Zhuang团队研发了基于单分子成像的方法MERFISH,即Multiplexed Error-Robust Fluorescence In Situ Hybridization,通过设计能检测纠错的编码方案,在单细胞分辨率下对千种RNA成像。
技术原理
将选择好的基因制成编码探针,也就是把barcodes(Error-robust)分配给细胞的RNA,通过组合标签(combinatorial labeling)、纠错编码(error-robust barcoding)、顺序成像(sequencitial imaging)三个核心步骤,结合标识细胞位置的染色结果,在单细胞水平上鉴定数千种RNA的拷贝数和空间定位,实现空间转录组表达测量。
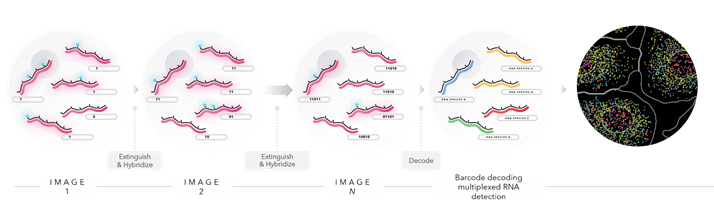
技术思路:如何基于smFISH,提升通量?
顺序成像
前期研究引入了mRNA编码。尽管smFISH后续被扩展到允许多种荧光染料同时对几种mRNA成像,通量依然受到限制。由于光谱可区分染料的数量远远小于细胞中活跃基因数量,无法完成每增加一种mRNA就增加一种染料以区分不同mRNAs。除了用不同颜色的组合以区分mRNA,使用杂交次序(hybridization rounds)的组合也能对不同mRNAs编码,即使用同一位置不同成像轮次的荧光信号编码mRNAs
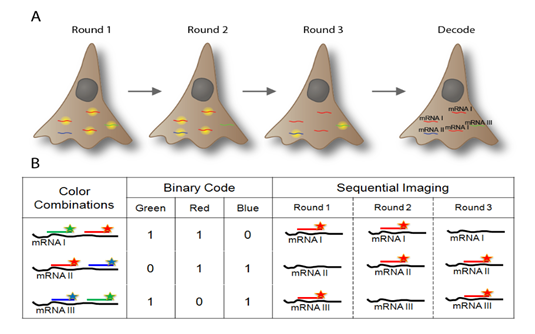
但顺序成像并非完美方案,主要面临以下问题:
- 随着测序rounds (N)的增加,标注(labing) 多种RNA需要长实验时间和大量荧光探针,合成花费高。
- 随着测序rounds (N)的增加,由于每轮杂交过程中检测信号的错误累积,导致检测效率下降和错误识别增加。
针对以上问题,MERFISH团队采用了以下两个方法:
大规模合成寡核苷酸探针(针对问题1)
MERFISH使用先前已开发出一种 Oligopaint 方法,用于从包含数万个定制序列的阵列衍生(array-derived)寡核苷酸池(oligo-pools)中生成大量寡核苷酸探针,以标记染色体 DNA 。
除了使用大量编码探针提高效率的同时,由于在初级杂交步骤中仅标记RNA一次,有效降低了探针合成成本。具体而言,与read-out序列杂交仅需15min,与细胞RNA杂交需要10h+。
组合标注&可纠错编码(针对问题2)
-
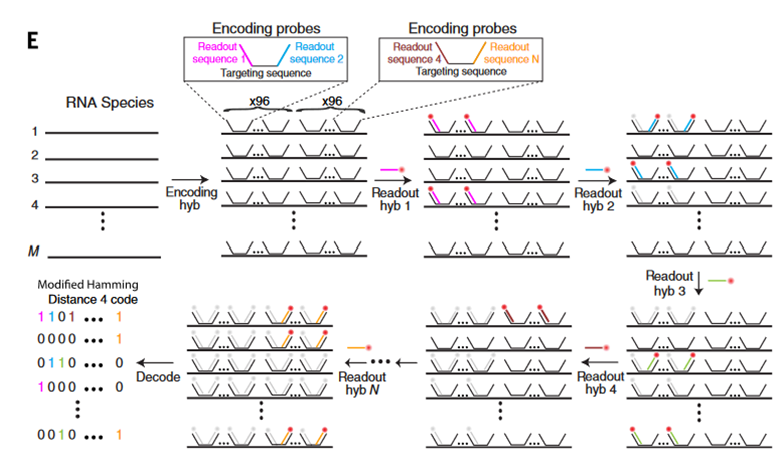
组合标注 组合标注:对每个RNA用~192个编码探针(encoding probe)标记。每个探针各自包括一个中央RNA靶向区域,两段侧翼是read-out序列。这些探针将RNA转换为特殊组合的read-out序列。特定RNA的编码探针包括4个N read-out特定序列组合,对应于该RNA应被读取到”1”的4轮杂交。N轮杂交之间,结合探针通过光漂白失活。
-
Error-robust barcodes Error-robust编码:统计结果表明,calling 错误(1→0)的比率(per bit)为10%,而错误识别(misidentification,0→1)的错误比率为4%。组合标记允许可检测的RNA物种的数量随着成像轮数呈指数增长,但检测误差也呈指数级增长。MERFISH基于扩展的汉明距离(d=4,MHD4) 设计Barcodes,为每个RNA分配一个二进制码,并根据二进制码用readout序列组合对RNA编码。
在MHD4中,位数上为”1”bits的数目恒定为4,两个code words之间间隔d=4汉明距离。如果存在一位读数错误(1 bit),可纠错将读数分配给最近的正确Barcode;如果存在两位读数错误(2 bits),能够检测出错误而无法纠正。

应用
- Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH doi:*https://doi.org/0.1126/science.abm1741
概述:使用MERFISH实现了对100多种神经元和非神经元细胞群的原位识别,绘制了具有高空间分辨率的人类大脑颞上回(superior temporal gyrus,STG)和颞中回(middle temporal gyrus,MTG)细胞图谱,探讨了细胞空间组织模式在人类和小鼠之间的异同
- Comparative analysis of MERFISH spatial transcriptomics with bulk and single-cell RNA sequencing MERFISH空间转录组学与批量scRNA-seq的比较分析 doi:*https://doi.org/10.1101/2022.03.04.483068
概述:文章对小鼠肝脏和肾脏组织同时进行了MERFISH (307-gene pannel)和bulk RNA-seq,scRNA-seq分析,给出结论:相比于scRNA-seq,MERFISH提供了定量可比的方法测量具有完整空间信息的单细胞基因表达,且能够独立解析肝脏肾脏的细胞亚型与空间结构(不需要集成scRNA-seq分析信息)。
个人思考
Key points
Pros
- MERFISH比bulk RNA-seq检测出更多转录本(10x – 1000x);
- MERFISH比scRNA-seq具有更高的灵敏度(scRNA-seq扭曲了免疫细胞计数,MERFISH可产生更准确的RNA统计数据);
- MERFISH能够单独有效分辨出不同细胞亚群(resolve between different subpopulations),结果与scVI/scANVI自动注释结果对比,表明不需要其他RNA-seq信息补充
Cons
- 影响信号质量最主要因素是细胞分割。MERFISH使用了DAPI+膜蛋白抗体的组合用以荧光识别细胞核、膜边界 (MERlin 图像分析)。只有少部分(30-50%)转录本被有效存留(分配给细胞),大量的细胞在前期处理就被过滤。
- 对于少数转录本数量极高的细胞(即每个细胞超过1000个),由于RNA分子过于拥挤,MERFISH图像中的smFISH点会过于密集,无法准确识别单个RNA转录本。
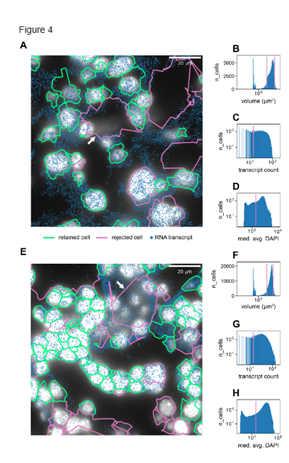 图. MERFISH分割错误与QC
图. MERFISH分割错误与QC
Key notes
- Limited probe panel of Marker genes。MERFISH需要探针设计,因此先验的scRNA-seq信息是必要的,用来评估MERFISH结果是否包括足够的信息量。
- 尽管根据peer review文章中显示MERFISH已能分辨空间结构与细胞亚型信息,但是需要关注到其现有局限性:对于表达丰度高的基因,由于细胞荧光信号拥挤效应(molecular crowding effect)导致检测偏差;同时细胞分割引入的误差也值得注意。
Reference
-
[MERFISH Spatial Profiling Technology Vizgen](https://vizgen.com/technology/) -
[Spatially resolved, highly multiplexed RNA profiling in single cells Science](https://www.science.org/doi/10.1126/science.aaa6090) -
[Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH Science](https://www.science.org/doi/10.1126/science.abm1741)